
Igalmi
- Generic: dexmedetomidine
- Dosage Form sublingual film, for sublingual or buccal use
Medically reviewed by A Ras MD. Last updated April 8, 2022
What is Igalmi
IGALMI is used to treat agitation in persons who have schizophrenia or bipolar I or II illness in adults
1 INDICATIONS AND USAGE
IGALMI is indicated for the acute treatment of agitation associated with schizophrenia or bipolar I or II disorder in adults.
Limitations of Use
The safety and effectiveness of IGALMI have not been established beyond 24 hours from the first dose [see Warnings and Precautions (5.4, 5.5)].
2 DOSAGE AND ADMINISTRATION
2.1 Important Recommendations Prior to Initiating IGALMI and During Therapy
IGALMI should be administered under the supervision of a healthcare provider. A healthcare provider should monitor vital signs and alertness after IGALMI administration to prevent falls and syncope [see Warnings and Precautions (5.5)].
IGALMI is for sublingual or buccal administration. Do not chew or swallow IGALMI. Do not eat or drink for at least 15 minutes after sublingual administration, or at least one hour after buccal administration.
2.2 Recommended Dosage
Table 1 includes dosage recommendations for IGALMI based on agitation severity for adults, patients with hepatic impairment, and geriatric patients. Lower dosages are recommended for patients with hepatic impairment and geriatric patients [see Warnings and Precautions (5.1) and Use in Specific Populations (8.5, 8.6)].
If agitation persists after the initial dose, up to two additional doses may be administered at least two hours apart. The dosage recommendations for additional doses vary depending upon the patient population and agitation severity (see Table 1). Assess vital signs including orthostatic measurements prior to the administration of any subsequent doses.
Due to risk of hypotension, additional half-doses are not recommended in patients with systolic blood pressure (SBP) less than 90 mmHg, diastolic blood pressure (DBP) less than 60 mmHg, heart rate (HR) less than 60 beats per minute, or postural decrease in SBP ≥ 20 mmHg or in DBP ≥ 10 mmHg.
Table 1: Dosage Recommendations for IGALMI in Adults, Adult Patients with Hepatic Impairment, and Geriatric Patients with Agitation Associated with Schizophrenia or Bipolar I or II Disorder
| Patient Population | Agitation Severity | Initial
Dose* |
Optional 2nd/3rd Doses* | Maximum
Recommended Total Daily Dosage |
| Adults | Mild or Moderate | 120 mcg | 60 mcg | 240 mcg |
| Severe | 180 mcg | 90 mcg | 360 mcg | |
| Patients with Mild or
Moderate Hepatic Impairment** |
Mild or Moderate | 90 mcg | 60 mcg | 210 mcg |
| Severe | 120 mcg | 60 mcg | 240 mcg | |
| Patients with Severe
Hepatic Impairment** |
Mild or Moderate | 60 mcg | 60 mcg | 180 mcg |
| Severe | 90 mcg | 60 mcg | 210 mcg | |
| Geriatric Patients
(≥ 65 years old) |
Mild, Moderate, or Severe | 120 mcg | 60 mcg | 240 mcg |
* IGALMI 120 mcg and 180 mcg dosage strengths may be cut in half to obtain the 60 mcg and 90 mcg doses, respectively [see Dosage and Administration (2.3)].
** Hepatic impairment: Mild (Child-Pugh Class A); Moderate (Child-Pugh Class B); Severe (Child-Pugh Class C)
2.3 Preparation and Administration Instructions
Keep IGALMI in the foil pouch until ready to administer. IGALMI should be immediately administered once the pouch is opened and the dose prepared.
Prepare and administer IGALMI under the supervision of a healthcare provider as follows:
| Healthcare Professional: Prepare IGALMI Dose for Patient | ||
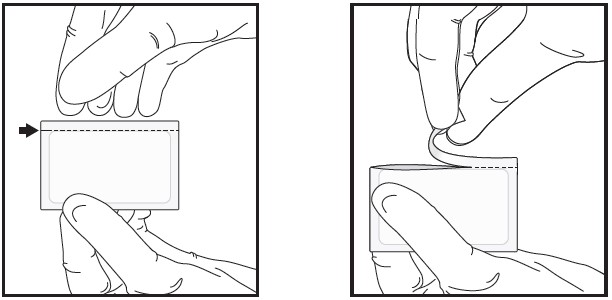
| 1 | Open the sealed foil pouch by tearing straight across at the notch.
|
 |
| • Perform Steps 2a, 2b, 2c and 2d only if a 60 mcg or 90 mcg dose (half of a film) is needed, then proceed to Step 3.
• If administering a full dose (1 film), proceed directly to Step 3.
|
||
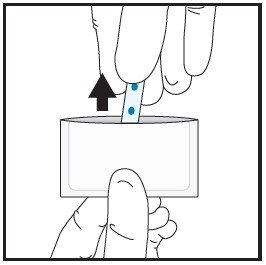
| 2a | Remove the film from the pouch with clean dry hands.
|
 |
| 2b | Cut the film in half between the dots with clean, dry scissors.
|
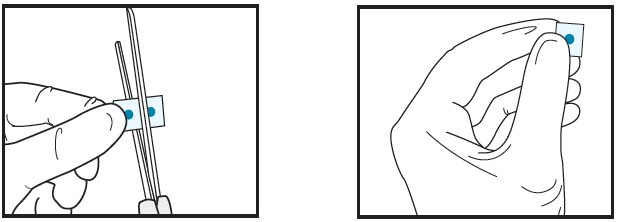 |
| 2c | Discard unused half in waste container.
|
|
| 2d | Place the half film for
administration to the patient back into the pouch. |
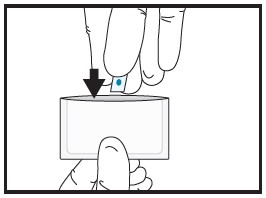 |
| 3 | Immediately give the pouch to the patient. | 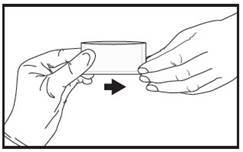 |
| 4 | Instruct patient to remove the film from the pouch with clean dry hands. | 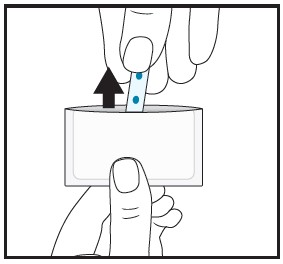 |
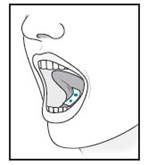
| 5 | For sublingual administration: Instruct patient to place film under the tongue. The film will stick in place.
Note: Patient may not eat or drink for 15 minutes after sublingual administration.
|
 |
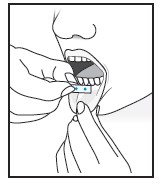
| For buccal administration: Instruct patient to place film behind lower lip. The film will stick in place.
Note: Patient may not eat or drink for one hour after buccal administration. |
 |
|
| 6 | Instruct patient to:
• Close their mouth. • Allow the film to dissolve. • Do not chew or swallow the film. |
|
3 DOSAGE FORMS AND STRENGTHS
IGALMI is a blue rectangular sublingual film containing on its surface two darker blue spots in dose strengths of 120 mcg and 180 mcg.
4 CONTRAINDICATIONS
None.
5 WARNINGS AND PRECAUTIONS
5.1 Hypotension, Orthostatic Hypotension, and Bradycardia
IGALMI causes dose-dependent hypotension, orthostatic hypotension, and bradycardia. In clinical studies, 18%, 16%, and 9% of patients treated with 180 mcg of IGALMI, 120 mcg of IGALMI, and placebo, respectively, experienced orthostatic hypotension (defined as SBP decrease ≥ 20 mmHg or DBP decrease ≥ 10 mmHg after 1, 3, or 5 minutes of standing) at 2 hours post-dose. In those studies, 7%, 6%, and 1% of patients treated with 180 mcg of IGALMI, 120 mcg of IGALMI, and placebo, respectively, experienced HR ≤ 50 beats per minute within 2 hours of dosing [see Adverse Reactions (6.1)]. In clinical studies with IGALMI, patients were excluded if they had treatment with alpha-1 noradrenergic blockers, benzodiazepines, other hypnotics or antipsychotic drugs four hours prior to study drug administration; had a history of syncope or syncopal attacks; SBP < 110 mmHg; DBP < 70 mmHg; HR < 55 beats per minute; or had evidence of hypovolemia or orthostatic hypotension.
Reports of hypotension and bradycardia, including some resulting in fatalities, have been associated with the use of another dexmedetomidine product given intravenously (IGALMI is for sublingual or buccal use and is not approved for intravenous use). Clinically significant episodes of bradycardia and sinus arrest have been reported after administration of this other dexmedetomidine product to young, healthy adult volunteers with high vagal tone and when this product was given by rapid intravenous or bolus administration.
Because IGALMI decreases sympathetic nervous system activity, hypotension and/or bradycardia may be more pronounced in patients with hypovolemia, diabetes mellitus, or chronic hypertension, and in geriatric patients [see Dosage and Administration (2.2) and Use in Specific Populations (8.5)].¶
Avoid use of IGALMI in patients with hypotension, orthostatic hypotension, advanced heart block, severe ventricular dysfunction, or history of syncope. After IGALMI administration, patients should be adequately hydrated and should sit or lie down until vital signs are within normal range. If a patient is unable to remain seated or lying down, precautions should be taken to reduce the risk of falls. Ensure that a patient is alert and not experiencing orthostatic hypotension or symptomatic hypotension prior to allowing them to resume ambulation [see Dosage and Administration (2.1)].
5.2 QT Interval Prolongation
IGALMI prolongs the QT interval. Avoid use of IGALMI in patients at risk of torsades de pointes or sudden death including those with known QT prolongation, a history of other arrhythmias, symptomatic bradycardia, hypokalemia or hypomagnesemia, and in patients receiving other drugs known to prolong the QT interval [see Drug Interactions (7.1) and Clinical Pharmacology (12.2)].
5.3 Somnolence
IGALMI can cause somnolence. In placebo-controlled clinical studies in adults with agitation associated with schizophrenia or bipolar I or II disorder, somnolence (including fatigue and sluggishness) was reported in 23% and 22% of patients treated with IGALMI 180 mcg and 120 mcg, respectively, compared to 6% of placebo-treated patients. Patients should not perform activities requiring mental alertness, such as operating a motor vehicle or operating hazardous machinery, for at least eight hours after taking IGALMI [see Adverse Reactions (6.1)].
5.4 Risk of Withdrawal Reactions
Symptoms of withdrawal have been observed after procedural sedation with another dexmedetomidine product administered intravenously. In this study, 12 (5%) adult patients who received intravenous dexmedetomidine up to 7 days (regardless of dose) experienced at least 1 event related to withdrawal within the first 24 hours after discontinuing dexmedetomidine and 7 (3%) adult patients who received intravenous dexmedetomidine experienced at least 1 event related with withdrawal 24 to 48 hours after discontinuing dexmedetomidine. The most common withdrawal reactions were nausea, vomiting, and agitation. In these subjects, tachycardia and hypertension requiring intervention occurred at a frequency of <5% in the 48 hours following intravenous dexmedetomidine discontinuation.
IGALMI was not studied for longer than 24 hours after the first dose. There may be a risk of physical dependence and a withdrawal syndrome if IGALMI is used in a manner other than indicated [see Dosage and Administration (2.2) and Drug Abuse and Dependence (9.3)].
5.5 Tolerance and Tachyphylaxis
Use of another dexmedetomidine product administered intravenously beyond 24 hours has been associated with tolerance and tachyphylaxis and a dose-related increase in adverse reactions.
IGALMI was not studied for longer than 24 hours after the first dose. There may be a risk of tolerance and tachyphylaxis if IGALMI is used in a manner other than indicated [see Dosage and Administration (2.2) and Drug Abuse and Dependence (9.3)].
6 ADVERSE REACTIONS
The following adverse reactions are discussed in detail in other sections of the labeling:
- Hypotension, Orthostatic Hypotension, and Bradycardia [see Warnings and Precautions (1)]
- QT Interval Prolongation [see Warnings and Precautions (2)]
- Somnolence [see Warnings and Precautions (3)]
- Risk of Withdrawal Reactions [see Warnings and Precautions (4)]
- Tolerance and Tachyphylaxis [see Warnings and Precautions (5)]
6.1 Clinical Studies Experience
Because clinical trials are conducted under widely varying conditions, adverse reactions rates observed in the clinical trials of a drug cannot be directly compared to rates in clinical trials of another drug and may not reflect the rates observed in practice.
The safety of IGALMI was evaluated in 507 adult patients with agitation associated with schizophrenia (N=255) or bipolar I or II disorder (N=252) in two randomized, placebo-controlled studies (Studies 1 and 2) [see Clinical Studies (14)]. In both studies, patients were admitted to a clinical research unit or a hospital and remained under medical supervision for at least 24 hours following treatment. Patients were 18 to 71 years of age (mean age was 46 years old); 45% were female and 55% were male; 66% were Black, 31% were White, 2% were multiracial, and 1% were other.
In these studies, patients received an initial dose of IGALMI 180 mcg (N=252), IGALMI 120 mcg (N=255), or placebo (N=252). Patients who were hemodynamically stable (i.e., those with systolic blood pressure (SBP) > 90 mmHg, diastolic blood pressure (DBP) > 60 mmHg, and heart rate (HR) > 60 beats per minute) and without orthostatic hypotension (i.e., reduction in SBP < 20 mmHg or DBP < 10 mmHg upon standing) were eligible for an additional dose after 2 hours. An additional half dose (90 mcg, 60 mcg, or placebo) was given to 7.1% (18/252), 22.7% (58/255) and 44.0% (111/252) of patients in the IGALMI 180 mcg, IGALMI 120 mcg or placebo arms, respectively. After at least an additional 2 hours, an additional second half dose (total IGALMI dose of 360 mcg, total IGALMI dose of 240 mcg, or placebo, respectively) was given to 3.2% (8/252), 9.4% (24/255), and 21.0% (53/252) of patients in the IGALMI 180 mcg, IGALMI 120 mcg or placebo arms, respectively.
In these studies, one patient discontinued treatment due to an adverse reaction of oropharyngeal pain. The most common adverse reactions (incidence ≥ 5% and at least twice the rate of placebo) were: somnolence, paresthesia or oral hypoesthesia, dizziness, dry mouth, hypotension, and orthostatic hypotension.
Table 2 presents the adverse reactions that occurred in IGALMI-treated patients at a rate of at least 2% and at a higher rate than in placebo-treated patients in Studies 1 and 2.
Table 2: Adverse Reactions Reported in ≥2% of IGALMI-Treated Patients and Greater than Placebo in Two Placebo-Controlled Studies of Agitated Adult Patients with Schizophrenia or Bipolar I or II Disorder (Studies 1 and 2)
| Adverse Reaction | IGALMI 180 mcg
N=252 % |
IGALMI 120 mcg
N=255 % |
Placebo
N=252 % |
| Somnolence1 | 23 | 22 | 6 |
| Paresthesia or hypoesthesia oral | 7 | 6 | 1 |
| Dizziness | 6 | 4 | 1 |
| Hypotension | 5 | 5 | 0 |
| Orthostatic hypotension | 5 | 3 | <1 |
| Dry mouth | 4 | 7 | 1 |
| Nausea | 3 | 2 | 2 |
| Bradycardia | 2 | 2 | 0 |
| Abdominal discomfort2 | 2 | 0 | 1 |
1Somnolence includes the terms fatigue and sluggishness
2Abdominal discomfort includes dyspepsia, gastroesophageal reflux disease
Hypotension, Orthostatic Hypotension, and Bradycardia in Two Placebo-Controlled Studies In clinical studies, patients were excluded if they were treated with alpha-1 noradrenergic blockers, benzodiazepines, antipsychotic drugs, or other hypnotics four hours prior to study drug administration; had a history of syncope or syncopal attacks; their SBP was less than 110 mmHg; their DBP was less than 70 mmHg; their HR was less than 55 beats per minute; or they had evidence of hypovolemia or orthostatic hypotension. In these studies, vital signs were monitored (at 30 minutes, 1-, 2-, 4-, 6-, and 8hours post-dose), including orthostatic vital signs at 2-, 4-, and 8-hours post-dose. Maximum positional decreases in SBP and DBP after standing were observed at two hours post-dose. Maximal reductions on BP and HR were observed two hours post-dose.
Table 3 presents the mean BP and HR decrease across all patients from both studies at 2 hours post dose.
Table 3: Mean Blood Pressure and Heart Rate Decrease At 2 Hours Post-Dose
| IGALMI 180 mcg N=252 | IGALMI 120 mcg N=255 | Placebo N=252 | |
| Mean SBP Decrease (mmHg) | 15 | 13 | 1 |
| Mean DBP Decrease (mmHg) | 8 | 7 | <1 |
| Mean Heart Rate Decrease (bpm) | 9 | 7 | 3 |
In the clinical studies:
- 13%, 8%, and < 1% of patients in the single dose 180 mcg IGALMI, 120 mcg IGALMI, and placebo groups, respectively, experienced SBP ≤ 90 mmHg and a decrease ≥ 20 mmHg of SBP within 24 hours of dosing.
- 19%, 17%, and 2% of the patients in the 180 mcg IGALMI, 120 mcg IGALMI, and placebo groups, respectively, had a DBP ≤ 60 mmHg and a DBP decrease ≥ 10 mmHg within 24 hours of dosing.
- 4%, 3%, and 0% of patients in the 180 mcg IGALMI, 120 mcg IGALMI, and placebo groups, respectively, had a HR ≤ 50 beats per minute and a HR decrease ≥ 20 beats per minute within 24 hours of dosing.
At 8 hours post-dose, 2% of patients in the IGALMI 180 mcg group experienced a SBP ≤ 90 mmHg and decrease ≥ 20 mmHg compared with one patient (<1%) in the IGALMI 120 mcg group and none in the placebo group. At 24 hours, none of the patients in the IGALMI 180 mcg group experienced a SBP ≤90 mmHg and decrease ≥ 20 mmHg compared with one patient (<1%) in the IGALMI 120 mcg group and none in the placebo group. At 8 hours post-dose, none of the patients in the IGALMI 180 mcg group had a HR ≤ 50 beats per minute and a HR decrease ≥ 20 beats per minute compared with one patient in the 120 mcg group (<1%) and none in the placebo group.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post approval use of another
dexmedetomidine product given intravenously (IGALMI is not approved for intravenous use). Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Blood and Lymphatic System Disorders: Anemia
- Cardiac Disorders: Arrhythmia, atrial fibrillation, atrioventricular block, bradycardia, cardiac arrest, cardiac disorder, extrasystoles, myocardial infarction, supraventricular tachycardia, tachycardia, ventricular arrhythmia, ventricular tachycardia
- Eye Disorders: Photopsia, visual impairment
- Gastrointestinal Disorders: Abdominal pain, diarrhea, nausea, vomiting
- General Disorders and Administration Site Conditions: Chills, hyperpyrexia, pain, pyrexia, thirst
- Hepatobiliary Disorders: Hepatic function abnormal, hyperbilirubinemia
- Investigations: Alanine aminotransferase increased, aspartate aminotransferase increased, blood alkaline phosphatase increased, blood urea increased, electrocardiogram T wave inversion, gammaglutamyltransferase increased, electrocardiogram QT prolonged
- Metabolism and Nutrition Disorders: Acidosis, hyperkalemia, hypoglycemia, hypovolemia, hypernatremia
- Nervous System Disorders: Convulsion, dizziness, headache, neuralgia, neuritis, speech disorder
- Psychiatric Disorders: Agitation, confusional state, delirium, hallucination, illusion
- Renal and Urinary Disorders: Oliguria, polyuria
- Respiratory, Thoracic and Mediastinal Disorders: Apnea, bronchospasm, dyspnea, hypercapnia, hypoventilation, hypoxia, pulmonary congestion, respiratory acidosis
- Skin and Subcutaneous Tissue Disorders: Hyperhidrosis, pruritus, rash, urticaria
- Surgical and Medical Procedures: Light anesthesia
- Vascular Disorders: Blood pressure fluctuation, hemorrhage, hypertension, hypotension
7 DRUG INTERACTIONS
7.1 Drugs that Prolong the QT Interval
Concomitant use of drugs that prolong the QT interval may add to the QT-prolonging effects of IGALMI and increase the risk of cardiac arrhythmia. Avoid the use of IGALMI in combination with other drugs known to prolong the QT interval [see Warnings and Precautions (5.2)].
7.2 Anesthetics, Sedatives, Hypnotics, and Opioids
Concomitant use of IGALMI with anesthetics, sedatives, hypnotics, or opioids is likely to lead to enhanced CNS depressant effects. Specific studies with another dexmedetomidine product given intravenously have confirmed these effects with sevoflurane, isoflurane, propofol, alfentanil, and midazolam. Due to possible enhanced CNS effects when given concomitantly with IGALMI, consider a reduction in dosage of IGALMI or the concomitant anesthetic, sedative, hypnotic, or opioid.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available data on IGALMI use in pregnant women to evaluate for a drug-associated risk of major birth defects, miscarriage or other adverse maternal or fetal effects. Available data from published randomized controlled trials and case reports over several decades of use with intravenously administered dexmedetomidine during pregnancy have not identified a drug-associated risk of major birth defects or miscarriage; however, the reported exposures occurred after the first trimester. Most of the available data are based on studies with exposures that occurred at the time of cesarean-section delivery, and these studies have not identified an adverse effect on maternal outcomes or infant Apgar scores. Available data indicate that dexmedetomidine crosses the placenta.
In animal reproductive studies fetal toxicity occurred in the presence of maternal toxicity with subcutaneous administration of dexmedetomidine to pregnant rats during organogenesis at doses 5 times the maximum recommended human dose [MRHD] of 360 mcg/day based on mg/m2 body surface area. Adverse developmental effects, including early implantation loss and decreased viability of second generation offspring, occurred when pregnant rats were subcutaneously administered doses less than or equal to the MRHD based on mg/m2 from late pregnancy through lactation and weaning (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Animal Data
Increased post-implantation losses and reduced live pups in the presence of maternal toxicity (decreased body weight) occurred in a rat embryo-fetal development study in which pregnant dams were administered subcutaneous doses of dexmedetomidine of 200 mcg/kg/day (equivalent to 5 times the MRHD of 360 mcg/day based on mg/m2) during the period of organogenesis (Gestation Day (GD) 5 to 16). No embryo-fetal toxicity was observed at 20 mcg/kg/day (less than the MRHD of 360 mcg/day based on mg/m2). No malformations were reported at any dose level.
No malformation or embryo-fetal toxicity were observed in a rabbit embryo-fetal developmental study in which pregnant dams were administered dexmedetomidine intravenously at doses up to 96 mcg/kg/day (equivalent to 5 times the MRHD of 360 mcg/day based on mg/m2) during the period of organogenesis (GD 6 to 18).
Reduced pup and adult offspring weights and grip strength were reported in a rat developmental toxicology study in which pregnant females were administered dexmedetomidine subcutaneously at 8 mcg/kg/day (less than the MRHD of 360 mcg/day based on mg/m2) during late pregnancy through lactation and weaning (GD 16 to postnatal day [PND] 25). Decreased viability of second generation offspring and an increase in early implantation loss along with delayed motor development occurred at 32 mcg/kg/day (equivalent to the MRHD of 360 mcg/day based on mg/m2) when first generation offspring were mated. This study limited dosing to hard palate closure (GD 15-18) through weaning instead of standard dosing from implantation (GD 6-7) to weaning (PND 21).
8.2 Lactation
Risk Summary
Available published literature report the presence of dexmedetomidine in human milk following intravenous administration. There is no information regarding the effects of dexmedetomidine on the breastfed child or the effects on milk production. Advise women to monitor the breastfed infant for irritability. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for IGALMI and any potential adverse effects on the breastfed child from IGALMI or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of IGALMI have not been established in pediatric patients.
8.5 Geriatric Use
Fifteen geriatric patients (≥ 65 years of age) were enrolled (no patients were 75 years of age and older) in the clinical studies for acute treatment of agitation associated with schizophrenia or bipolar I or II disorder. Of the total number of IGALMI-treated patients in these clinical studies, 11/507 (2.2%) were 65 years of age and older [see Clinical Studies (14)].
Dosage reduction of IGALMI is recommended in geriatric patients [see Dosage and Administration (2.2)]. A higher incidence of bradycardia and hypotension was observed in geriatric patients compared to younger adult patients after intravenous administration of another dexmedetomidine product [see Warnings and Precautions (5.1)]. The pharmacokinetic profile of intravenous dexmedetomidine was not altered in geriatric subjects [see Clinical Pharmacology (12.3)].
Clinical studies of IGALMI did not include sufficient numbers of patients 65 years of age and older to determine whether there were differences in the effectiveness of IGALMI in the acute treatment of agitation associated with schizophrenia or bipolar I or II disorder compared to younger adult patients.
8.6 Hepatic Impairment
Dexmedetomidine clearance was decreased in patients with hepatic impairment (Child-Pugh Class A, B, or C). Thus, a dosage reduction of IGALMI is recommended in patients with hepatic impairment compared to patients with normal hepatic function [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)].
9 DRUG ABUSE AND DEPENDENCE
9.1 Controlled Substance
IGALMI contains dexmedetomidine, which is not a controlled substance.
9.3 Dependence
Physical Dependence
Physical dependence is a state that develops as a result of physiological adaptation in response to repeated drug use, manifested by withdrawal signs and symptoms after abrupt discontinuation or a significant dose reduction of a drug. The dependence potential of dexmedetomidine has not been studied in humans. However, because studies in rodents and primates have demonstrated that intravenous dexmedetomidine exhibits pharmacologic actions similar to those of clonidine, it is possible that dexmedetomidine may produce a clonidine-like withdrawal syndrome upon abrupt discontinuation. IGALMI was not studied for longer than 24 hours after the first dose. There may be risk of physical dependence and a withdrawal syndrome if IGALMI is used in a manner other than indicated [see Dosage and Administration (2.1, 2.2) and Warnings and Precautions (5.4)].
Tolerance
Tolerance is a physiological state characterized by a reduced response to a drug after repeated administration (i.e., a higher dose of a drug is required to produce the same effect that was once obtained at a lower dose).
IGALMI has not been studied for longer than 24 hours after the first dose. There may be a risk for tolerance if IGALMI is administered in a manner other than indicated [see Dosage and Administration (2.1, 2.2) and Warnings and Precautions (5.5)].
10 OVERDOSAGE
In a tolerability study of intravenous dexmedetomidine in which healthy adult subjects were administered doses at and above the recommended dose of 0.2 to 0.7 mcg/kg/hour, the maximum blood concentration was approximately 13 times the upper boundary of the therapeutic range for the intravenous dexmedetomidine (IGALMI is not approved for intravenous use). The most notable effects observed in two subjects who achieved the highest doses were first degree atrioventricular block and second-degree heart block.
Five adult patients received an overdose of intravenous dexmedetomidine in intensive care unit sedation studies. Two patients who received a 2 mcg/kg loading dose (twice the recommended loading dose) over 10 minutes, experienced bradycardia and/or hypotension.
One patient who received a loading intravenous bolus dose of undiluted dexmedetomidine (19.4 mcg/kg), had cardiac arrest from which he was successfully resuscitated.
Consider contacting a Poison Center (1-800-222-1222) or a medical toxicologist for overdosage management recommendations for IGALMI.
11 DESCRIPTION
IGALMI contains dexmedetomidine, an alpha2-adrenergic receptor agonist, present as dexmedetomidine hydrochloride, the S-enantiomer of medetomidine chemically described as 4-[(1S)-1(2, 3-dimethylphenyl) ethyl]-1H-imidazole hydrochloride. The empirical formula is C13H16N2•HCl with a molecular weight of 236.7 g/mol. The structural formula of dexmedetomidine hydrochloride is: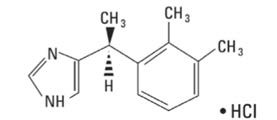
Dexmedetomidine hydrochloride is a white or almost white powder that is freely soluble in water and has a pKa of 7.1. Its partition coefficient in octanol/water at pH 7.4 is 2.89.
IGALMI is for sublingual or buccal use. Each IGALMI sublingual film contains 120 mcg or 180 mcg of dexmedetomidine equivalent to 141.8 mcg and 212.7 mcg of dexmedetomidine hydrochloride, respectively.
IGALMI contains the following inactive ingredients: FD&C Blue #1 colorant, hydroxypropyl cellulose, peppermint oil, polyethylene oxide, and sucralose.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Dexmedetomidine is an alpha-2 adrenergic receptor agonist. The mechanism of action of IGALMI in the acute treatment of agitation associated with schizophrenia or bipolar I or II disorder is thought to be due to activation of presynaptic alpha-2 adrenergic receptors.
12.2 Pharmacodynamics
Dexmedetomidine acts as an agonist at alpha-2 adrenergic receptors with binding affinities (Ki values) of 4 to 6 nM at the alpha-2 adrenergic receptor subtypes.
Cardiac Electrophysiology
IGALMI exhibits a concentration dependent QT prolongation. Table 4 shows the mean (upper 90% confidence interval) QTcF increase from baseline for respective dosing regimens [see Warnings and Precautions (5.2)].
Table 4: QTcF Increase from Baseline by Dosage of IGALMI
| IGALMI Dosage | Mean QTcF Increase from Baseline (upper 90% confidence interval) |
| 120 mcg single use | 6 (7) msec |
| 120 mcg + 2 additional doses of 60 mcg 2 hours apart (total 3 doses) | 8 (9) msec |
| 180 mcg single use | 8 (11) msec |
| 180 mcg + 2 additional doses of 90 mcg 2 hours apart (total 3 doses) | 11 (14) msec |
12.3 Pharmacokinetics
Dexmedetomidine exposure (Cmax and AUC) increased in a dose proportional manner in the dose range of 20 mcg (0.17 times the lowest recommended initial dose of 120 mcg) to 180 mcg after single sublingual administration of IGALMI.
The mean time for film to dissolve in the mouth was about 6 to 8 minutes and 18 minutes following sublingual and buccal administration, respectively. Dexmedetomidine was quantifiable in plasma generally after 5 to 20 minutes post dosing.
Absorption
The absolute bioavailability of dexmedetomidine was about 72% and 82% following sublingual and buccal administration of IGALMI, respectively. When water was taken at two hours post dose, comparable exposures of dexmedetomidine were observed when IGALMI was administered by both routes.
Mean maximal plasma concentrations of dexmedetomidine were reached approximately two hours after sublingual or buccal administration of IGALMI. Following sublingual administration of 40 mcg of IGALMI (0.33 times the lowest recommended initial dose) with water drinking at two hours post dose and 20 mcg dexmedetomidine intravenous infusion for 90 minutes in healthy volunteers:
- The mean peak plasma concentration (Cmax) of dexmedetomidine was 143 ng/L and 144 ng/L, respectively.
- The mean area under concentration curve (AUC) of dexmedetomidine was 851 hour*ng/L and 584 hour*ng/L, respectively.
Effect of Drinking Water on Absorption
Compared to drinking water at two hours post sublingual administration of IGALMI, early water intake
(as early as 15 minutes post dose) had minimal effects on the rate or extent of absorption of dexmedetomidine.
Effects of early water intake (i.e., before two hours post dose) on the absorption of dexmedetomidine has not been evaluated following buccal administration.
Distribution
The steady-state volume of distribution (Vss) of dexmedetomidine following intravenous administration was approximately 118 liters. Dexmedetomidine protein binding was assessed in the plasma of healthy male and female subjects. The average protein binding was 94% and was constant across the different plasma concentrations tested. Protein binding was similar in males and females. The fraction of dexmedetomidine that was bound to plasma proteins was significantly decreased in subjects with hepatic impairment compared to healthy subjects.
The potential for protein binding displacement of dexmedetomidine by fentanyl, ketorolac, theophylline, digoxin and lidocaine was explored in vitro, and negligible changes in the plasma protein binding of dexmedetomidine IV were observed. The potential for protein binding displacement of phenytoin, warfarin, ibuprofen, propranolol, theophylline and digoxin by dexmedetomidine hydrochloride injection was explored in vitro and none of these compounds appeared to be significantly displaced by intravenous dexmedetomidine.
Elimination
Metabolism
Dexmedetomidine undergoes almost complete biotransformation with very little unchanged dexmedetomidine excreted in urine and feces. Biotransformation involves both direct glucuronidation as well as cytochrome P450 mediated metabolism. The major metabolic pathways of dexmedetomidine are: direct N-glucuronidation to inactive metabolites; aliphatic hydroxylation (mediated primarily by CYP2A6 with a minor role of CYP1A2, CYP2E1, CYP2D6 and CYP2C19) of dexmedetomidine to generate 3-hydroxy-dexmedetomidine, the glucuronide of 3-hydroxy-dexmedetomidine, and 3-carboxydexmedetomidine; and N-methylation of dexmedetomidine to generate 3-hydroxy N-methyldexmedetomidine, 3-carboxy N-methyl-dexmedetomidine, and dexmedetomidine-N-methyl Oglucuronide.
Excretion
The mean terminal elimination half-life (t1/2) of dexmedetomidine is approximately 2.8 hours following sublingual or buccal administration of IGALMI. Clearance is estimated to be approximately 39 L/h following intravenous administration.
A mass balance study demonstrated that after nine days, an average of 95% of the radioactivity, following intravenous administration of radiolabeled dexmedetomidine, was recovered in the urine and 4% in the feces. No unchanged dexmedetomidine was detected in the urine. Approximately 85% of the radioactivity recovered in the urine was excreted within 24 hours after the infusion. Fractionation of the radioactivity excreted in urine demonstrated that products of N-glucuronidation accounted for approximately 34% of the cumulative urinary excretion. In addition, aliphatic hydroxylation of parent drug to form 3-hydroxy-dexmedetomidine, the glucuronide of 3-hydroxy-dexmedetomidine, and 3carboxylic acid-dexmedetomidine together represented approximately 14% of the dose in urine. Nmethylation of dexmedetomidine to form 3-hydroxy N-methyl dexmedetomidine, 3-carboxy N-methyl dexmedetomidine, and N-methyl O-glucuronide dexmedetomidine accounted for approximately 18% of the dose in urine. The N-methyl metabolite itself was a minor circulating component and was undetected in urine. Approximately 28% of the urinary metabolites have not been identified.
Specific Populations
Male and Female Patients
There was no observed difference in the pharmacokinetics of intravenous dexmedetomidine due to sex.
Geriatric Patients
The pharmacokinetic profile of intravenous dexmedetomidine was not altered by age. There were no differences in the pharmacokinetics of intravenous dexmedetomidine in young (18–40 years), middle age (41–65 years), and geriatric (>65 years) subjects.
Patients with Hepatic Impairment
In subjects with varying degrees of hepatic impairment (Child-Pugh Class A, B, or C), clearance values for intravenous dexmedetomidine were lower than in subjects with normal hepatic function [see Dosage and Administration (2.2)]. After an intravenous infusion of 0.6 mcg/kg of this dexmedetomidine product over 10 minutes the mean clearance values for subjects with mild, moderate, and severe hepatic impairment were 74%, 64% and 53% of those observed in subjects with normal hepatic function, respectively. Mean clearances for free drug were 59%, 51% and 32% of those observed in subjects with normal hepatic function, respectively.
Patients with Renal Impairment
Dexmedetomidine pharmacokinetics (Cmax, Tmax, AUC, t1/2, CL, and V) were not significantly different in patients with creatinine clearance <30 mL/minute compared to subjects with normal renal function.
Drug Interactions Studies
In vitro studies in human liver microsomes demonstrated no evidence of cytochrome P450 mediated drug interactions that are likely to be of clinical relevance.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Animal carcinogenicity studies have not been performed with dexmedetomidine.
Mutagenesis
Dexmedetomidine was not mutagenic in the in vitro Ames bacterial reverse mutation test or mammalian mouse lymphoma cell forward mutation assay. Dexmedetomidine was not clastogenic in the in vitro human lymphocyte chromosome aberration test in the absence or presence of human liver S9 metabolic activation, however, a weak clastogenic response was noted in the presence of rat liver S9 metabolic activation. Dexmedetomidine was not clastogenic in the in vivo bone marrow micronucleus test in CD-1 mice, although there was some evidence for clastogenicity in NMRI mice.
Impairment of Fertility
Fertility in male or female rats was not affected after daily subcutaneous injections of dexmedetomidine at doses up to 54 mcg/kg (1.5 times the MRHD of 360 mcg/day on a mg/m2 basis) administered from 10 weeks prior to mating in males, and 3 weeks prior to mating and during mating in females.
13.2 Animal Toxicology and/or Pharmacology
Twice daily sublingual administration of 120 to 320 mcg/day of dexmedetomidine to dogs for 28 days caused decreased heart rate and moderate sedation up to 3.5 hours post dose. A single male dog (out of 32 treated dogs) dosed 320 mcg/day (equivalent to the MRHD of 360 mcg/day) exhibited inflammation, necrosis, myofiber degeneration, and hemorrhage at the sublingual treatment site. No adverse effects were noted at 240 mcg/day (less than the MRHD of 360 mcg/day).
14 CLINICAL STUDIES
The effectiveness of IGALMI for the acute treatment of agitation associated with schizophrenia or bipolar I or II disorder in adults was established in two randomized, double-blind, placebo-controlled, fixed-dose studies (Studies 1 and 2):
- Study 1 (NCT04268303) included 380 patients who met DSM-5 criteria for schizophrenia, schizoaffective or schizophreniform disorder. The population was 18 to 71 years of age (mean age was 46 years old); 37% female and 63% male; 78% Black, 20% White, 1% multiracial, and 1% Asian.
- Study 2 (NCT04276883) included 378 patients who met DSM-5 criteria for bipolar I or II disorder. The population was 18 to 70 years of age (mean age was 47 years old); 45% female and 55% male; 56% Black, 41% White, 1% Asian, 1% multiracial, and 1% other.
The Positive and Negative Syndrome Scale-Excited Component (PEC) is an investigator-rated instrument consisting of 5 items: poor impulse control, tension, hostility, uncooperativeness, and excitement. Each item is scored on a scale from 1 to 7 (1=absent, 2=minimal, 3=mild, 4=moderate, 5=moderate-severe, 6=severe, 7=extremely severe). The total PEC score ranges from 5 to 35, with higher scores reflecting greater overall symptom severity. For enrollment in the studies, patients had to be judged to be clinically agitated with a total PEC score of ≥ 14, with at least one individual item score ≥ 4. In both studies, patients were admitted to a clinical research unit or a hospital and remained under medical supervision for at least 24 hours following treatment.
Patients were randomized to receive a single sublingual dose of 180 mcg of IGALMI, 120 mcg of
IGALMI, or placebo. The primary efficacy endpoint in both studies was the change from baseline in the
PEC score, assessed two hours following the initial dose. The key secondary endpoint was the time to effect onset, assessed by measuring the change from baseline in PEC score at 10, 20, 30, 45, 60, and 90 minutes after the initial dose administration.
In both studies, mean baseline PEC scores were similar in all treatment groups (Table 5). The mean change from baseline in the PEC total score at two hours after the first dose in patients treated with 180 mcg and 120 mcg of IGALMI was statistically greater than patients who received placebo (Table 5). Examination of population subsets (race and sex) on the primary endpoint did not show evidence for differential responsiveness between White and Black or female and male patients. The clinical studies did not include enough patients of other races or patients ≥65 years of age to determine whether there were differences in effectiveness for those groups.
Table 5: Primary Efficacy Results for Change from Baseline in the PEC Score at Two Hours in Agitated Patients with Schizophrenia or Bipolar I or II Disorder (Studies 1 and 2)
| Study | Treatment Group | Number of
Patients |
Mean
Baseline PEC Score (SD) |
LS Mean
Change from Baseline to 2 hour Post First Dose (SE) |
LS Mean
Difference (95% CI) |
| Study 1 | IGALMI 180 mcg* | 125 | 17.6 (2.7) | -10.3 (0.4) | -5.5 (-6.5, -4.4) |
| IGALMI 120 mcg* | 129 | 17.5 (2.5) | -8.5 (0.4) | -3.7 (-4.8, -2.7) | |
| Placebo | 126 | 17.6 (2.3) | -4.8 (0.4) | – | |
| Study 2 | IGALMI 180 mcg* | 126 | 18.0 (3.0) | -10.4 (0.4) | -5.4 (-6.5, -4.3) |
| IGALMI 120 mcg* | 126 | 18.0 (2.7) | -9.1 (0.4) | -4.1 (-5.1, -3.0) | |
| Placebo | 126 | 17.9 (2.9) | -5.0 (0.4) | – | |
| SD=standard deviation; SE=standard error; LS Mean=least-squares mean; CI=unadjusted confidence interval; PEC=Positive and Negative Syndrome
Scale-Excited Component * IGALMI doses that were statistically significantly superior to placebo after adjusting for multiplicity. |
|||||
Figures 1 and 2 show the changes from baseline in PEC score at each time point assessed up to two hours following the initial dose. In Study 1, the decrease in agitation with IGALMI, compared to placebo, was statistically significant beginning at 20 minutes following dosing with the 180 mcg dose and 30 minutes after the 120 mcg dose. In Study 2, the decrease in agitation with IGALMI, compared to placebo, was statistically significant beginning at 20 minutes after treatment with both 120 mcg and 180 mcg doses.
Figure 1: Mean Change from Baseline in PEC Score Through Two Hours after a Single Dose in Agitated Patients with Schizophrenia (Study 1)
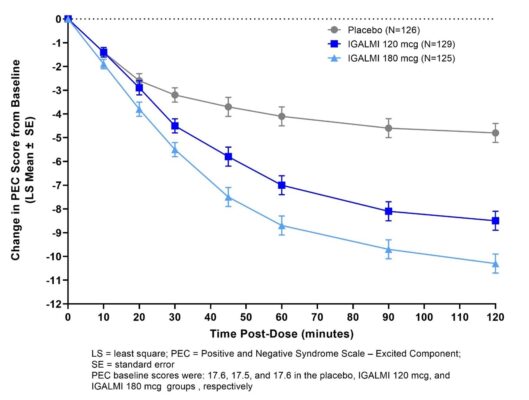
Figure 2: Mean Change from Baseline in PEC Score Through Two Hours after a Single Dose in
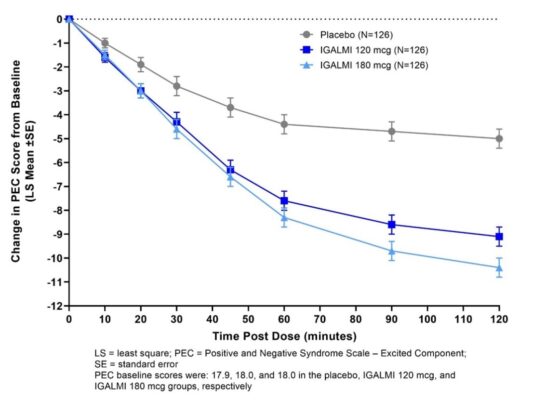
Agitated Patients with Bipolar I or II Disorder (Study 2)
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
IGALMI (dexmedetomidine) sublingual film is supplied as a blue rectangular sublingual film, containing on its surface two darker blue spots in dose strengths of 120 mcg and 180 mcg and is packaged as individual films in heat-sealed foil pouches in 10-count and 30-count films per carton. The NDC number for each packaging configuration is:
- 120 mcg 10-count NDC # 81092-1120-1
- 120 mcg 30-count NDC # 81092-1120-3
- 180 mcg 10-count NDC # 81092-1180-1
- 180 mcg 30-count NDC # 81092-1180-3
Storage and Handling
Store at controlled room temperature, 20°C to 25°C (68°F to 77°F). Excursions permitted from 15°C to 30°C (59°F to 86°F). See USP Controlled Room Temperature.
Keep IGALMI in the foil pouch until ready to administer.
17 PATIENT COUNSELING INFORMATION
Administration Information
Advise patients to place IGALMI under the tongue, close to the base of the tongue, on the left or right side (sublingual) or behind the lower lip (buccal).
Advise patients not to chew or swallow IGALMI. Also, advise patients not to eat or drink for at least 15 minutes after sublingual administration, or at least 1 hour after buccal administration [see Dosage and Administration (2.3)].
Hypotension, Orthostatic Hypotension, and Bradycardia
Advise patients that IGALMI can cause dose-dependent hypotension, orthostatic hypotension, and bradycardia. Inform patients to remain sitting or lying down after receiving IGALMI and to inform the healthcare provider if they have any symptoms of hypotension or bradycardia [see Warnings and Precautions (5.1)].
QT Interval Prolongation
Inform patients to consult their physician immediately if they feel faint or have heart palpitations [see Warnings and Precautions (5.2)].
Somnolence
Advise patients that IGALMI can cause somnolence and may impair the ability to perform tasks that require complex motor and mental skills. Advise patients that they should avoid doing activities that require them to be alert, such as driving a car or operating machinery for at least eight hours after receiving IGALMI [see Warnings and Precautions (5.3)].
Lactation
Advise patients exposed to IGALMI to monitor breastfed infants for irritability [see Use in Specific Populations (8.2)]